solo per uso di ricerca
MK-8617 HIF modulatore
N. Cat.S8443
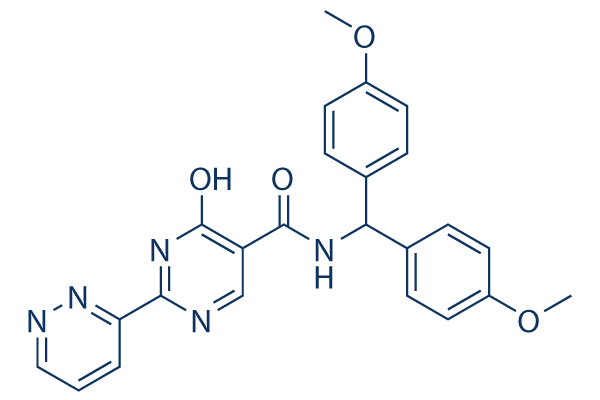
Struttura chimica
Peso molecolare: 443.45
Controllo Qualità
| Target correlati | EGFR VEGFR JAK PDGFR FGFR Src FLT FLT3 HER2 Bcr-Abl |
|---|---|
| Altro HIF Inibitori | PT2399 PX-478 Dihydrochloride BAY 87-2243 KC7F2 Lificiguat (YC-1) IOX2 CAY10585 (LW 6) Molidustat (BAY 85-3934) PT2385 IDF-11774 |
Informazioni chimiche, conservazione e stabilità
| Peso molecolare | 443.45 | Formula | C24H21N5O4 |
Conservazione (Dalla data di ricezione) | |
|---|---|---|---|---|---|
| N. CAS | 1187990-87-9 | Scarica SDF | Conservazione delle soluzioni stock |
|
|
| Sinonimi | N/A | Smiles | COC1=CC=C(C=C1)C(C2=CC=C(C=C2)OC)NC(=O)C3=CN=C(NC3=O)C4=NN=CC=C4 | ||
Solubilità
|
In vitro |
DMSO
: 10 mg/mL
(22.55 mM)
Water : Insoluble Ethanol : Insoluble |
Calcolatore di Molarità
|
In vivo |
|||||
Calcolatore di formulazione in vivo (Soluzione chiara)
Passo 1: Inserire le informazioni di seguito (Consigliato: Un animale aggiuntivo per tenere conto della perdita durante lesperimento)
Passo 2: Inserire la formulazione in vivo (Questo è solo il calcolatore, non la formulazione. Contattateci prima se non cè una formulazione in vivo nella sezione Solubilità.)
Risultati del calcolo:
Concentrazione di lavoro: mg/ml;
Metodo per preparare il liquido master di DMSO: mg farmaco predissolto in μL DMSO ( Concentrazione del liquido master mg/mL, Vi preghiamo di contattarci prima se la concentrazione supera la solubilità del DMSO del lotto del farmaco. )
Metodo per preparare la formulazione in vivo: Prendere μL DMSO liquido master, quindi aggiungereμL PEG300, mescolare e chiarire, quindi aggiungereμL Tween 80, mescolare e chiarire, quindi aggiungere μL ddH2O, mescolare e chiarire.
Metodo per preparare la formulazione in vivo: Prendere μL DMSO liquido master, quindi aggiungere μL Olio di mais, mescolare e chiarire.
Nota: 1. Si prega di assicurarsi che il liquido sia limpido prima di aggiungere il solvente successivo.
2. Assicurarsi di aggiungere il/i solvente/i in ordine. È necessario assicurarsi che la soluzione ottenuta, nellaggiunta precedente, sia una soluzione limpida prima di procedere allaggiunta del solvente successivo. Metodi fisici come il vortex, gli ultrasuoni o il bagno dacqua calda possono essere utilizzati per facilitare la dissoluzione.
Meccanismo dazione
| Targets/IC50/Ki |
PHD1
(Cell-free assay) 1 nM
PHD2
(Cell-free assay) 1 nM
PHD3
(Cell-free assay) 14 nM
|
|---|---|
| In vitro |
MK-8617 non è un inibitore significativo degli enzimi del citocromo p450 in vitro (IC50), CYP1A2, 3A4, 2B6, 2C9, 2C19 o 2D6, >60 μM, ed è un inibitore reversibile moderato di CYP2C8 a 1,6 μM in vitro. Questo composto è inattivo se testato a 10 μM contro un pannello generale di 171 saggi di legame con radioligandi e saggi enzimatici. |
| In vivo |
Il MK-8617 tritiato mostra un minimo turnover metabolico nei microsomi epatici (+NADPH) di ratto, cane e scimmia (<10% di turnover) ma un significativo turnover nei microsomi epatici umani (34% di turnover) dopo 60 minuti (10 μM di composto, 1 mg/mL di proteina microsomiale). In termini di profilo farmacocinetico, questo composto mostra una buona biodisponibilità orale in tutte le specie (36−71%), con bassa clearance e volume di distribuzione. Il composto ha ancora un'emivita di eliminazione relativamente lunga nelle specie precliniche. Nei topi (C57Bl/6), singole dosi di 5 e 15 mpk po (n = 3) causano aumenti dei reticolociti circolanti misurati sia al 3° che al 4° giorno dopo la somministrazione del composto. Nel ratto (Sprague−Dawley), una titolazione di dose singola di 1,5, 5 e 15 mpk po (n = 5) causa un grande aumento dei livelli sierici di eritropoietina (EPO) di 1,7-, 8- e 204 volte rispetto al veicolo, rispettivamente. Aumenti dei reticolociti circolanti sono osservati a 5 e 15 mg/kg 3 giorni dopo la somministrazione e, con la dose di 15 mg, 4 giorni dopo la somministrazione. |
Riferimenti |
|
Supporto tecnico
Istruzioni per la manipolazione
Tel: +1-832-582-8158 Ext:3
Per qualsiasi altra domanda, si prega di lasciare un messaggio.
I prodotti sono solo per uso di ricerca. Non per uso umano. Non vendiamo a pazienti.
©Copyright 2013 Selleck Chemicals. Tutti i diritti riservati.