solo per uso di ricerca
Fimasartan Angiotensin Receptor antagonista
N. Cat.S4975
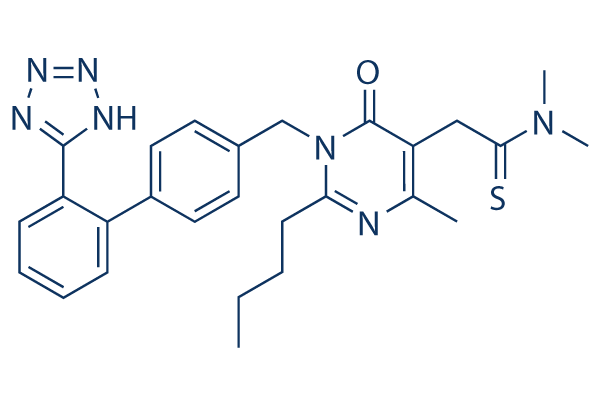
Struttura chimica
Peso molecolare: 501.65
Controllo Qualità
- Citato in Nature Medicine per la sua qualità superiore
- COA
- SDS
- Scheda tecnica
| Target correlati | CXCR Hedgehog/Smoothened PKA Adrenergic Receptor AChR 5-HT Receptor Histamine Receptor Dopamine Receptor Ras KRas |
|---|---|
| Altro Angiotensin Receptor Inibitori | PD123319 ML221 A-779 Olodanrigan (EMA401) Buloxibutid AVE 0991 |
Informazioni chimiche, conservazione e stabilità
| Peso molecolare | 501.65 | Formula | C27H31N7OS |
Conservazione (Dalla data di ricezione) | |
|---|---|---|---|---|---|
| N. CAS | 247257-48-3 | -- | Conservazione delle soluzioni stock |
|
|
| Sinonimi | Kanarb | Smiles | CCCCC1=NC(=C(C(=O)N1CC2=CC=C(C=C2)C3=CC=CC=C3C4=NNN=N4)CC(=S)N(C)C)C | ||
Solubilità
|
In vitro |
DMSO
: 100 mg/mL
(199.34 mM)
Ethanol : 24 mg/mL Water : Insoluble |
Calcolatore di Molarità
|
In vivo |
|||||
Calcolatore di formulazione in vivo (Soluzione chiara)
Passo 1: Inserire le informazioni di seguito (Consigliato: Un animale aggiuntivo per tenere conto della perdita durante lesperimento)
Passo 2: Inserire la formulazione in vivo (Questo è solo il calcolatore, non la formulazione. Contattateci prima se non cè una formulazione in vivo nella sezione Solubilità.)
Risultati del calcolo:
Concentrazione di lavoro: mg/ml;
Metodo per preparare il liquido master di DMSO: mg farmaco predissolto in μL DMSO ( Concentrazione del liquido master mg/mL, Vi preghiamo di contattarci prima se la concentrazione supera la solubilità del DMSO del lotto del farmaco. )
Metodo per preparare la formulazione in vivo: Prendere μL DMSO liquido master, quindi aggiungereμL PEG300, mescolare e chiarire, quindi aggiungereμL Tween 80, mescolare e chiarire, quindi aggiungere μL ddH2O, mescolare e chiarire.
Metodo per preparare la formulazione in vivo: Prendere μL DMSO liquido master, quindi aggiungere μL Olio di mais, mescolare e chiarire.
Nota: 1. Si prega di assicurarsi che il liquido sia limpido prima di aggiungere il solvente successivo.
2. Assicurarsi di aggiungere il/i solvente/i in ordine. È necessario assicurarsi che la soluzione ottenuta, nellaggiunta precedente, sia una soluzione limpida prima di procedere allaggiunta del solvente successivo. Metodi fisici come il vortex, gli ultrasuoni o il bagno dacqua calda possono essere utilizzati per facilitare la dissoluzione.
Meccanismo dazione
| Targets/IC50/Ki |
AT1 receptor
|
|---|---|
| In vitro |
Fimasartan è un antagonista selettivo del recettore AT1. La concentrazione che inibisce il legame di [125I]Ang II al recettore AT1 della corteccia surrenale di ratto del 50% (IC50) è 0,13 nM. Questo composto attenua potentemente la morte apoptotica miocardica nel cuore di ratto riperfuso e nelle cellule H9c2. Potrebbe attenuare il danno mitocondriale associato alla minimizzazione della riduzione di Bcl-2 e collegato alla riduzione di p53 e all'attivazione di Akt e GSK-3β, e inibire il sovraccarico mitocondriale di Ca2+ sopprimendo l'ICa,L e MCU. |
| In vivo |
In vari modelli animali, inclusi ratti ipertesi renali, ratti spontaneamente ipertesi e cani Beagle, fimasartan riduce efficacemente la pressione sanguigna in modo dose-dipendente dopo somministrazione orale e endovenosa singola o ripetuta. Questo composto non influisce sul comportamento generale, sulla frequenza respiratoria o sul volume corrente negli animali sperimentali e non mostra risultati avversi nel test del gene hERG (human ether-a-go-go-related gene) o nello studio di telemetria sulle scimmie. Sono stati condotti numerosi studi di tossicità generale, tossicità carcinogenica, tossicità genetica e tossicità dello sviluppo somministrati per via orale o endovenosa in varie specie, inclusi topi, ratti, scimmie e cani, e questi risultati preclinici dimostrano la sicurezza e la tollerabilità di questa sostanza chimica per l'uso clinico a lungo termine e offrono margini di sicurezza sufficienti per supportare la dose terapeutica umana attesa. Viene rapidamente assorbito dopo somministrazione orale con un tempo al picco di concentrazione plasmatica (Tmax) che varia da 0,5 a 3 ore e un'emivita terminale (t1/2) di 5-16 ore a dosi da 20 a 480 mg in soggetti sani. Risultati simili si ottengono in pazienti con ipertensione, cioè il Tmax varia da 0,5 a 1,3 ore e la t1/2 è di 7-10 ore dopo la somministrazione di questo composto a dosi di 20-180 mg nello studio di fase II successivo. L'escrezione urinaria di questa sostanza chimica è bassa, con l'escrezione urinaria complessiva del farmaco immodificato nelle prime 24 ore dopo la somministrazione inferiore al 3% della dose somministrata. Subisce eliminazione non renale con metabolismo minimo. Il trattamento con questo composto prima dell'ischemia miocardica attenua significativamente il danno da riperfusione e i cambiamenti apoptotici, possibilmente sopprimendo il danno mitocondriale durante la riperfusione. In vari studi preclinici, inclusi modelli di lesione da pallone aortico, ischemia/riperfusione da infarto miocardico, cardiotossicità da doxorubicina e ictus ischemico, questa sostanza chimica mostra effetti antinfiammatori e di protezione degli organi. |
Riferimenti |
|
Informazioni sullo studio clinico
(dati da https://clinicaltrials.gov, aggiornato il 2024-05-22)
| Numero NCT | Reclutamento | Condizioni | Sponsor/Collaboratori | Data di inizio | Fasi |
|---|---|---|---|---|---|
| NCT02994745 | Completed | Hypertension Hyperlipidemia |
Boryung Pharmaceutical Co. Ltd |
December 23 2016 | Phase 1 |
| NCT02920047 | Completed | Hypertension |
Boryung Pharmaceutical Co. Ltd |
October 2016 | Phase 1 |
| NCT02995720 | Completed | Hypertension Hyperlipidemia |
Boryung Pharmaceutical Co. Ltd |
August 26 2016 | Phase 1 |
| NCT03231293 | Completed | Hypertension|Ischemic Stroke|Transient Ischemic Attack |
Boryung Pharmaceutical Co. Ltd |
July 28 2016 | -- |
Supporto tecnico
Istruzioni per la manipolazione
Tel: +1-832-582-8158 Ext:3
Per qualsiasi altra domanda, si prega di lasciare un messaggio.
I prodotti sono solo per uso di ricerca. Non per uso umano. Non vendiamo a pazienti.
©Copyright 2013 Selleck Chemicals. Tutti i diritti riservati.