solo per uso di ricerca
Tariquidar (XR9576) P-gp inibitore
N. Cat.S8028
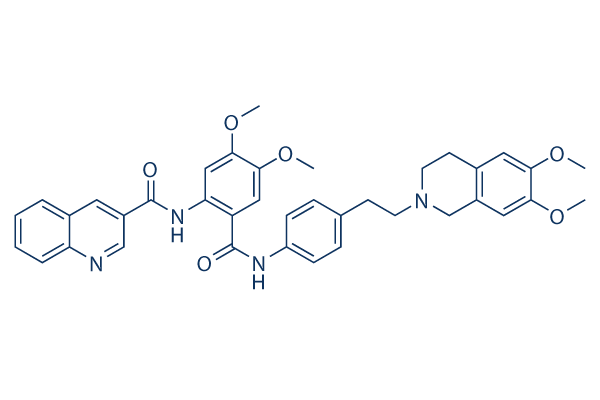
Struttura chimica
Peso molecolare: 646.73
Controllo Qualità
Coltura cellulare, trattamento e concentrazione di lavoro
| Linee cellulari | Tipo di saggio | Concentrazione | Tempo di incubazione | Formulazione | Descrizione dellattività | PMID |
|---|---|---|---|---|---|---|
| K562/DOX | Function assay | 1 uM | 10 mins | Inhibition of P-gp in human K562/DOX cells assessed as increase in rhodamine-123 efflux in human K562 cells at 1 uM incubated for 10 mins hrs by flow cytometry relative to untreated control | 28113128 | |
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| KB-V1 | Function assay | 200 nM | Inhibition of P-gp in human KB-V1 cells assessed as increase in rhodamine 123 accumulation at 200 nM | 21657271 | ||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| HepG2 | Cytotoxicity assay | 48 hrs | Cytotoxicity against adriamycin-resistant human HepG2 cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=37.2μM | 27328029 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells after 48 hrs by MTT assay, IC50=31.56μM | 28645831 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=31.56μM | 29631786 | ||
| A2780adr | Function assay | 10 uM | 30 mins | Inhibition of ABCB1 in human A2780adr cells assessed as increase in accumulation of calcein AM at 10 uM preincubated for 30 mins followed by calcein AM addition measured every 60 secs for 60 mins by fluorescence assay relative to control | 29547272 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells overexpressing P-gp assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=27.19μM | 29631786 | ||
| CCD-18Co | Cytotoxicity assay | 48 hrs | Cytotoxicity against human CCD-18Co cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| SW620/AD300 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620/AD300 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| HLF | Cytotoxicity assay | 48 hrs | Cytotoxicity against HLF cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=16.69μM | 27328029 | ||
| CEM/VLB500 | Growth inhibition assay | 3 days | Growth inhibition of human CEM/VLB500 cells after 3 days by resazurin assay, GI50=13.5μM | 17399990 | ||
| MCF7/ADR | Cytotoxicity assay | 48 hrs | Intrinsic cytotoxicity against human MCF7/ADR cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=13.1μM | 27328029 | ||
| HCT116 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human HCT116 cells assessed as cell viability after 48 hrs by MTT assay, IC50=12.5μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 12 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=8.28μM | 28645831 | ||
| KBV | Function assay | 5 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 5 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=5.24μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 6 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=4.97μM | 28645831 | ||
| KBV | Function assay | 10 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 10 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=4.46μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured immediately by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=3.02μM | 28645831 | ||
| K562/A02 | Function assay | 5 uM | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 at 5 uM measured after 48 hrs by MTT assay (Rvb = 43.75 to 96.91 uM), IC50=1.97μM | 28645831 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 measured after 48 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=1.6μM | 28645831 | ||
| MCF-7 MX | Function assay | Inhibition of BCRP expressed in MCF-7 MX cells using Hoechst 33342 staining, IC50=1.5μM | 21354800 | |||
| MCF7 | Function assay | Inhibition of ABCG2 in human mitoxantrone-resistant MCF7 cells by Hoechst 33342 assay, IC50=1.44544μM | 18678495 | |||
| HFE | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HFE cells assessed as cell viability after 72 hrs by MTT assay, IC50=1.28μM | 26197160 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells using Hoechst 33342 staining, IC50=0.94μM | 21354800 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 overexpressed in human MCF7/Topo cells by flow cytometric-based mitoxantrone efflux assay, IC50=0.916μM | 19170519 | |||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 staining based fluorescence assay, IC50=0.526μM | 30128080 | ||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 microplate assay, IC50=0.526μM | 24900683 | ||
| MCF7/Topo | Function assay | Inhibition of ABCG2 expressed in human MCF7/Topo cells by Hoechst microplate assay, IC50=0.526μM | 21570282 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 in human MCF7/Topo cells by Hoechst 33342 assay, IC50=0.52μM | 26774038 | |||
| KBV1 | Function assay | 10 mins | Inhibition of ABCB1 in human KBV1 cells after 10 mins by Calcein-AM microplate assay, IC50=0.223μM | 24900683 | ||
| Kb-V1 | Function assay | 10 mins | Inhibition of ABCB1 expressed in Kb-V1 cells after 10 mins by calcein-AM assay, IC50=0.223μM | 21570282 | ||
| KBv1 | Function assay | Inhibition of ABCB1 overexpressed in human KBv1 cells by flow cytometric-based calcein-AM efflux assay, IC50=0.223μM | 19170519 | |||
| KBV1 | Function assay | Inhibition of ABCB1 in human KBV1 cells assessed as inhibition of calcein-AM efflux, IC50=0.22μM | 26774038 | |||
| A2780 | Function assay | 30 mins | Inhibition of human Pgp in A2780 cells after 30 mins by Hoechst 33342 assay, IC50=0.12589μM | 18083034 | ||
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human KB-3-1 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.15 +/- 0.04 uM), IC50=0.11μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human OVCAR8 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.12 +/- 0.03 uM), IC50=0.08μM | 27504669 | |
| A2780/ADR | Function assay | Inhibition of P-glycoprotein-mediated multidrug resistance in adriamycin-resistant human A2780/ADR cells by calcein AM assay, IC50=0.078μM | 19250834 | |||
| A2780adr | Function assay | Inhibition of P-gp expressed in A2780adr cells by calcein AM accumulation assay, IC50=0.08μM | 21354800 | |||
| A2780 | Function assay | Inhibition of P-gp in human adriamycin-resistant A2780 cells by Hoechst 33342 assay, IC50=0.07244μM | 18678495 | |||
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.07 +/- 0.19 uM), IC50=0.07μM | 27504669 | |
| CEM/VLB500 | Function assay | 3 days | Reversal of P-gp-mediated multidrug resistance to in human CEM/VLB500 cells after 3 days by resazurin assay, EC50=0.068μM | 17399990 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.06457μM | 18083034 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.064μM | 18083034 | ||
| MDCK | Function assay | 30 mins | Inhibition of P-glycoprotein (unknown origin) expressed in MDCK cells assessed as reduction of calcein-AM transport after 30 mins by fluorescence assay, EC50=0.044μM | 24607999 | ||
| MDCK | Function assay | 30 mins | Activity at MDR1 (unknown origin) expressed in MDCK cells using calcein AM as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.044μM | 23374872 | ||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 3714.80 +/- 383.58 nM), IC50=0.01851μM | 27504669 | |
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 277.68 +/- 56.61 nM), IC50=0.00066μM | 27504669 | |
| HEK293 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against HEK293 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 5.28 +/- 0.74 nM), IC50=0.00495μM | 27504669 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells after 48 hrs by MTT assay, IC50=27.19μM | 28645831 | ||
| SW620 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 24 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=14.39μM | 28645831 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells by pheophorbide A assay, IC50=0.85μM | 19932960 | |||
| MCF7 MX | Function assay | Inhibition of BCRP expressed in MCF7 MX cells by Hoechst 33342 staining, IC50=0.68μM | 19932960 | |||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.54 +/- 0.60 uM), IC50=0.24μM | 27504669 | |
| MDCK | Function assay | Inhibition of MDR1 expressed in MDCK cells using rhodamine 123 staining by flow cytometry, IC50=0.21μM | 21354800 | |||
| CCRF-CEM/VCR1000 | Function assay | 240 secs | Inhibition of P-glycoprotein-mediated daunorubicin efflux from human CCRF-CEM/VCR1000 cells after 240 secs by FACS flow cytometric analysis, IC50=0.03311μM | 22452412 | ||
| HEK293 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 transfected in HEK293 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 504.65 +/- 44.94 nM), IC50=0.02477μM | 27504669 | |
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human KB-3-1 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.78 +/- 0.27 nM), IC50=0.00041μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human OVCAR8 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 8.53 +/- 1.95 nM), IC50=0.00518μM | 27504669 | |
| MDCK | Function assay | 30 mins | Activity at BCRP (unknown origin) expressed in MDCK cells using rhodamine 123 as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.01μM | 23374872 | ||
| HepG2 | Function assay | 10 uM | 90 mins | Inhibition of P-gp mediated efflux in adriamycin-resistant human HepG2 cells assessed as intracellular rhodamine-123 accumulation at 10 uM incubated in dark condition for 90 mins by flow cytometry relative to control | 27328029 | |
| Clicca per visualizzare più dati sperimentali sulle linee cellulari | ||||||
Informazioni chimiche, conservazione e stabilità
| Peso molecolare | 646.73 | Formula | C38H38N4O6 |
Conservazione (Dalla data di ricezione) | |
|---|---|---|---|---|---|
| N. CAS | 206873-63-4 | Scarica SDF | Conservazione delle soluzioni stock |
|
|
| Sinonimi | XR9576 | Smiles | COC1=C(C=C2CN(CCC2=C1)CCC3=CC=C(C=C3)NC(=O)C4=CC(=C(C=C4NC(=O)C5=CC6=CC=CC=C6N=C5)OC)OC)OC | ||
Solubilità
|
In vitro |
DMSO
: 8 mg/mL
(12.36 mM)
Water : Insoluble Ethanol : Insoluble |
Calcolatore di Molarità
|
In vivo |
|||||
Calcolatore di formulazione in vivo (Soluzione chiara)
Passo 1: Inserire le informazioni di seguito (Consigliato: Un animale aggiuntivo per tenere conto della perdita durante lesperimento)
Passo 2: Inserire la formulazione in vivo (Questo è solo il calcolatore, non la formulazione. Contattateci prima se non cè una formulazione in vivo nella sezione Solubilità.)
Risultati del calcolo:
Concentrazione di lavoro: mg/ml;
Metodo per preparare il liquido master di DMSO: mg farmaco predissolto in μL DMSO ( Concentrazione del liquido master mg/mL, Vi preghiamo di contattarci prima se la concentrazione supera la solubilità del DMSO del lotto del farmaco. )
Metodo per preparare la formulazione in vivo: Prendere μL DMSO liquido master, quindi aggiungereμL PEG300, mescolare e chiarire, quindi aggiungereμL Tween 80, mescolare e chiarire, quindi aggiungere μL ddH2O, mescolare e chiarire.
Metodo per preparare la formulazione in vivo: Prendere μL DMSO liquido master, quindi aggiungere μL Olio di mais, mescolare e chiarire.
Nota: 1. Si prega di assicurarsi che il liquido sia limpido prima di aggiungere il solvente successivo.
2. Assicurarsi di aggiungere il/i solvente/i in ordine. È necessario assicurarsi che la soluzione ottenuta, nellaggiunta precedente, sia una soluzione limpida prima di procedere allaggiunta del solvente successivo. Metodi fisici come il vortex, gli ultrasuoni o il bagno dacqua calda possono essere utilizzati per facilitare la dissoluzione.
Meccanismo dazione
| Targets/IC50/Ki |
P-gp
(CHrB30 cells) 5.1 nM(Kd)
|
|---|---|
| In vitro |
Tariquidar mostra un legame ad alta affinità con P-gp con un Bmax di 275 pmol/mg. Questo composto mostra un'interazione non competitiva con i substrati di P-gp. Aumenta l'accumulo allo stato stazionario di questi citotossici nelle cellule CHrB30 a livelli osservati nelle cellule AuxB1 non esprimenti P-gp con una EC50 di 487 nM. Questa sostanza chimica è in grado di inibire l'attività ATPasica vanadato-sensibile di P-gp del 60-70%, con potenti valori di IC50 di 43 nM. Può inibire altri meccanismi di resistenza a concentrazioni più elevate. 1 µM di questo composto abroga la resistenza mediata da ABCG2 (BCRP) alle camptotecine in vitro. Potenzia la citotossicità di diversi farmaci, inclusa la doxorubicina; il completo rovesciamento della resistenza è ottenuto in presenza di 25-80 nM di questa sostanza chimica. In MC26, una linea cellulare di carcinoma del colon murino con chemioresistenza intrinseca, l'IC50 della doxorubicina è cinque volte inferiore in presenza di 0,1 µM di questo composto (36 vs 7 nM). In linee cellulari di carcinoma mammario murino, carcinoma polmonare a piccole cellule umano e carcinoma ovarico umano con resistenza chemioterapica acquisita (EMT6/AR1.0, H69/LX4 e 2780 AD), l'IC50 della doxorubicina in vitro è 22-150 volte inferiore in presenza di 0,1 µM di questa sostanza chimica. L'inibizione di P-gp persiste per 23 ore dopo la sua rimozione dal sistema di coltura. Ha ripristinato la citotossicità della doxorubicina nel modello di sferoide tumorale multicellulare NCI/ADRRES (National Cancer Institute) derivato dalla linea cellulare di cancro al seno MCF7WT. |
| Saggio chinasico |
Saggio di accumulo del farmaco allo stato stazionario
|
|
Le cellule vengono incubate in un volume di reazione di 1 mL per 60 min a 37 ℃ sotto 5% CO2 al fine di raggiungere lo stato stazionario. L'effetto dei modulatori XR9576 sull'accumulo del ligando [3H] viene investigato nell'intervallo di concentrazione 10-9 - 10-6 M. Questo composto viene aggiunto da una soluzione madre di DMSO, fornendo una concentrazione finale di solvente dello 0,2 % (v/v). Dopo la raccolta delle cellule, il farmaco accumulato viene misurato mediante conteggio a scintillazione liquida e normalizzato per il contenuto proteico cellulare. I grafici della quantità accumulata in funzione della concentrazione del modulatore vengono adattati con l'equazione generale dose-risposta: Y={(a-b)/(1+(X/c)d)}+bDove: Y=risposta; a=risposta iniziale; b=risposta finale; c=concentrazione EC50; d=valore di pendenza; X=concentrazione del farmaco.
|
|
| In vivo |
Tariquidar (2-8 mg/kg p.o.) si è dimostrato in grado di potenziare significativamente l'attività antitumorale della doxorubicina (5 mg/kg, i.v.) contro il carcinoma del colon murino MC26 in vivo. Negli xenotrapianti di carcinoma umano, la co-somministrazione di questo composto (6-12 mg/kg p.o.) ha completamente ripristinato l'attività antitumorale contro due xenotrapianti tumorali umani MDR altamente resistenti (2780AD, H69/LX4) in topi nudi. |
Riferimenti |
|
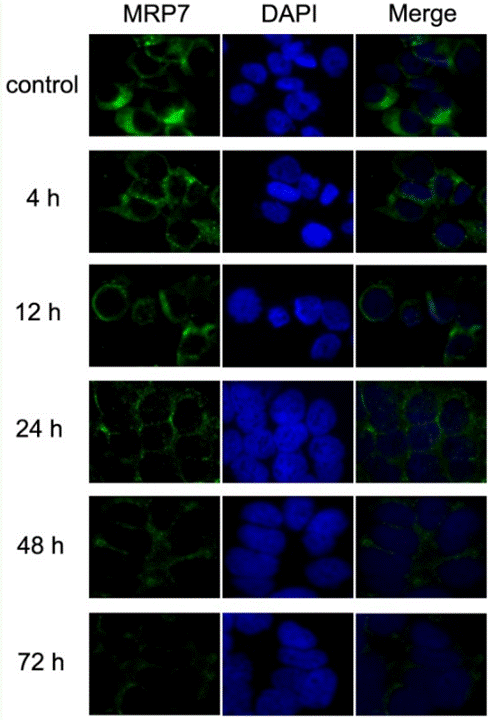
Applicazioni
| Metodi | Biomarcatori | Immagini | PMID |
|---|---|---|---|
| Immunofluorescence | MRP7 |
|
23393594 |
Informazioni sullo studio clinico
(dati da https://clinicaltrials.gov, aggiornato il 2024-05-22)
| Numero NCT | Reclutamento | Condizioni | Sponsor/Collaboratori | Data di inizio | Fasi |
|---|---|---|---|---|---|
| NCT01663545 | Completed | Epilepsies Partial |
National Institute of Neurological Disorders and Stroke (NINDS)|National Institutes of Health Clinical Center (CC) |
July 31 2012 | -- |
| NCT01547754 | Terminated | HIV-Associated Cognitive Motor Complex |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
January 9 2012 | -- |
| NCT01386476 | Completed | Drug Resistance |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
June 15 2011 | -- |
| NCT00082368 | Completed | Cancer |
National Cancer Institute (NCI)|National Institutes of Health Clinical Center (CC) |
May 16 2004 | Phase 2 |
Supporto tecnico
Istruzioni per la manipolazione
Tel: +1-832-582-8158 Ext:3
Per qualsiasi altra domanda, si prega di lasciare un messaggio.
Domande Frequenti
Domanda 1:
Can you please give me more specific and detailed information of how to dissolve and use it (S8028) for in vivo studies?
Risposta:
It in 30% Propylene glycol, 5% Tween 80, 65% D5W at 30mg/ml will be a suspension or emulsion. If you are going to administrate this compound by oral gavage, it is fine. We also have test some vehicles for it for i.p injection, and it is soluble in 5% DMSO+45% PEG 300+ddH2O at 2mg/ml clearly. When preparing the solution, please dissolve it in DMSO clearly first, then add PEG. After they mixed well, then dilute with water.
I prodotti sono solo per uso di ricerca. Non per uso umano. Non vendiamo a pazienti.
©Copyright 2013 Selleck Chemicals. Tutti i diritti riservati.