réservé à la recherche
Anlotinib (AL3818) Dihydrochloride Inhibiteur de VEGFR2
N° Cat.S8726
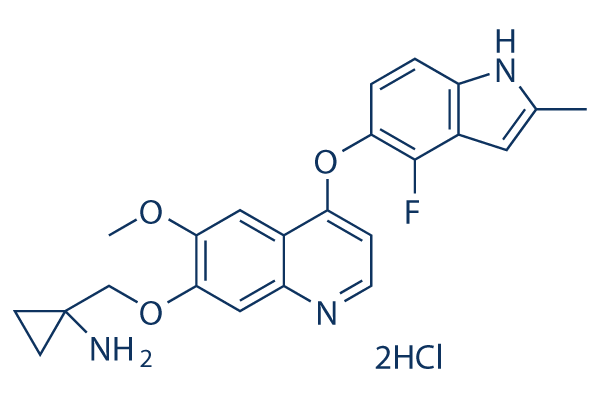
Structure chimique
Poids moléculaire: 480.36
Aller à
Contrôle qualité
Lot :
Pureté :
99.93%
99.93
| Cibles apparentées | EGFR PDGFR FGFR c-Met Src MEK CSF-1R FLT3 HER2 c-Kit |
|---|---|
| Autre VEGFR Inhibiteurs | SAR131675 SU 5402 Cediranib (AZD2171) Vatalanib (PTK787) 2HCl Linifanib (ABT-869) Apatinib (YN968D1) Apatinib (YN968D1) mesylate Ki8751 ZM 323881 HCl Semaxanib (SU5416) |
Culture cellulaire, traitement et concentration de travail
| Lignées cellulaires | Type dessai | Concentration | Temps dincubation | Formulation | Description de lactivité | PMID |
|---|---|---|---|---|---|---|
| U-87MG | Function assay | 0.01, 0.1, 1, 10 and 100 μM | 1.5 h | inhibited PDGF-BB-stimulated phosphorylation of PDGFRβ, AKT and ERK in U-87MG cells | 29446853 | |
| Mo7e | Function assay | 0.01, 0.1, 1, 10 and 100 μM | 1.5 h | Anlotinib inhibited SCF‐1‐stimulated phosphorylation of c‐Kit, AKT and ERK in Mo7e cells | 29446853 | |
| HUVEC | Function assay | 0.01, 0.1, 1, 10 and 100 μM | 1.5 h | anlotinib inhibited VEGF‐stimulated intracellular phosphorylation of VEGFR2 in a concentration‐dependent way in HUVEC with a subnanomolar IC50 value | 29446853 | |
| A549 | Cell viability assay | 24, 48 and 72 h | IC50=64.82 μM(t=24 h); IC50=30.34 μM(t=48 h); IC50=17.29 μM(t=72 h) | 30755242 | ||
| NCI-H1975 | Cell viability assay | 8 μg/ml | 24 h | Cell viability was decreased remarkably | 30871526 | |
| Calu-1 | Cell viability assay | 24, 48 and 72 h | IC50=61.23 μM(t=24 h); IC50=36.8 μM(t=48 h); IC50=28.64 μM(t=72 h) | 30755242 | ||
| Cliquez pour voir plus de données expérimentales sur les lignées cellulaires | ||||||
Informations chimiques, stockage et stabilité
| Poids moléculaire | 480.36 | Formule | C23H22FN3O3.2HCl |
Stockage (À partir de la date de réception) | 3 years -20°C powder |
|---|---|---|---|---|---|
| N° CAS | 1360460-82-7 | -- | Stockage des solutions mères |
|
|
| Synonymes | N/A | Smiles | CC1=CC2=C(N1)C=CC(=C2F)OC3=C4C=C(C(=CC4=NC=C3)OCC5(CC5)N)OC.Cl.Cl | ||
Solubilité
|
In vitro |
Water : 96 mg/mL
DMSO
: 48 mg/mL
(99.92 mM)
Ethanol : Insoluble |
Calculateur de molarité
Calculateur de dilution
Calculateur de poids moléculaire
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Entrez les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
mg/kg
g
μL
Étape 2 : Entrez la formulation in vivo (Ceci nest que le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
% DMSO
%
% Tween 80
% ddH2O
%DMSO
%
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuiteμL PEG300, mélanger et clarifier, ajouter ensuiteμL Tween 80, mélanger et clarifier, ajouter ensuite μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuite μL Huile de maïs, mélanger et clarifier.
Remarque : 1. Assurez-vous que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue lors de lajout précédent est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
VEGFR2
(Cell-free assay) 0.2 nM
VEGFR3
(Cell-free assay) 0.7 nM
c-Kit
(Cell-free assay) 14.8 nM
c-Kit
(Cell-free assay) 14.8 nM
c-Kit
(Cell-free assay) 14.8 nM
|
|---|---|
| In vitro |
L'Anlotinib occupe la poche de liaison de l'ATP de la tyrosine kinase VEGFR2 et montre une sélectivité et une puissance inhibitrice élevées (IC 50 <1 nmol/L) pour VEGFR2 par rapport à d'autres tyrosine kinases. L'Anlotinib inhibe VEGFR2 et VEGFR3 avec des valeurs d'IC50 de 0,2 et 0,7 nmol/L, respectivement. La puissance inhibitrice de l'anlotinib contre VEGFR1 est plus faible, avec une valeur d'IC50 de 26,9 nmol/L. Les valeurs d'IC50 de l'anlotinib pour l'inhibition des kinases liées au PDGFR c-Kit et PDGFRβ sont respectivement de 14,8 et 115,0 nmol/L. L'Anlotinib a peu d'effet sur l'activité d'autres kinases, y compris c-Met, c-Src, EGFR et HER2, même à une concentration de 2000 nmol/L. L'Anlotinib inhibe la signalisation induite par le VEGF et la prolifération cellulaire dans les HUVEC avec des valeurs d'IC50 picomolaires. Cependant, des concentrations micromolaires d'anlotinib sont nécessaires pour inhiber directement la prolifération des cellules tumorales in vitro. L'Anlotinib inhibe significativement la migration et la formation de tubes des HUVEC ; il inhibe également la croissance des microvaisseaux à partir d'explants d'aorte de rat in vitro.
|
| Kinase Assay |
Dosage immuno-enzymatique
|
|
L'activité inhibitrice de l'anlotinib contre les tyrosine kinases a été déterminée par ELISA. La réaction de l'ATP avec la tyrosine kinase a été initiée dans un tampon de réaction (50 mmol/L HEPES pH 7,4, 50 mmol/L MgCl2, 0,5 mmol/L MnCl2, 0,2 mmol/L Na3VO4, 1 mmol/L DTT) et incubée pendant 1 heure à 37°C dans des plaques à 96 puits pré-enduites de 20 μg/mL de Poly(Glu,Tyr)4:1. La plaque a été incubée avec l'anticorps PY99 puis avec l'IgG anti-souris conjuguée à la HRP. Après réaction avec une solution d'o-phénylènediamine puis arrêt avec l'ajout de 2N H2SO4, l'absorbance a été mesurée à 490 nm à l'aide d'un lecteur hybride Synergy H4.
|
|
| In vivo |
L'Anlotinib diminue la densité vasculaire dans les tissus tumoraux in vivo. Comparé au sunitinib, un inhibiteur de tyrosine kinase bien connu, une dose orale quotidienne d'anlotinib montre une efficacité antitumorale in vivo plus large et plus forte et, dans certains modèles, provoque une régression tumorale chez des souris nues. Il est bien toléré chez la souris. L'Anlotinib est efficace à des doses (1,5 à 6 mg/kg par jour) significativement inférieures aux doses efficaces d'autres TKI, qui nécessitent des doses de 20 à 100 mg/kg pour obtenir une inhibition significative de la croissance tumorale chez la souris. In vivo, l'anlotinib a montré une activité large contre des modèles de xénogreffes tumorales humaines du côlon (SW-620), de l'ovaire (SK-OV-3), du foie (SMMC-7721), du rein (Caki-1), du gliome (U87MG) et du poumon non à petites cellules (Calu-3) pendant la période de dosage. Chez les rats Sprague-Dawley et les chiens beagle, l'anlotinib est rapidement absorbé par le tractus gastro-intestinal après administration orale. La biodisponibilité orale est de 23 à 45 % chez les rats et de 47 à 74 % chez les chiens. L'Anlotinib présente un grand volume de distribution chez les deux espèces. Chez les rats, les tissus primaires, tels que le poumon, les reins, le foie et le cœur, présentent des niveaux d'exposition à l'anlotinib significativement plus élevés que ceux du plasma. Le niveau d'exposition dans le cerveau est comparable au niveau plasmatique correspondant. Chez les souris porteuses de tumeurs, l'anlotinib se concentre 2,4 à 2,6 fois plus dans le tissu tumoral que dans le plasma. Chez l'homme, l'anlotinib présente une t1/2 assez longue (96 ± 17 h), qui semblait être indépendante de la dose. La demi-vie terminale de l'anlotinib chez les chiens (22,8 ± 11,0 h) est plus longue que celle chez les rats (5,1 ± 1,6 h). Cette différence semble être principalement associée à une différence interspécifique de clairance plasmatique totale (rats, 5,35 ± 1,31 L/h/kg ; chiens, 0,40 ± 0,06 L/h/kg). Dans le plasma humain, l'anlotinib est principalement lié à l'albumine et aux lipoprotéines, plutôt qu'à l'α1-glycoprotéine acide ou aux γ-globulines.
|
Références |
|
Applications
| Méthodes | Biomarqueurs | Images | PMID |
|---|---|---|---|
| Western blot | TP53 / Cleaved-caspase 3 / Cleaved PARP Beclin-1 / LC3-I / LC3-II Akt / p-Akt / mTOR / p-mTOR |
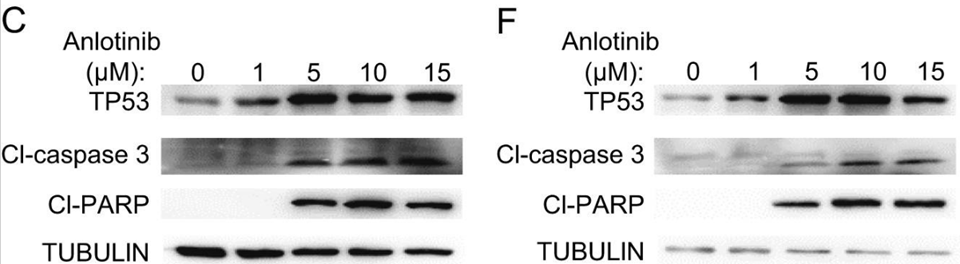
|
30139768 |
| Immunofluorescence | LC3-II |
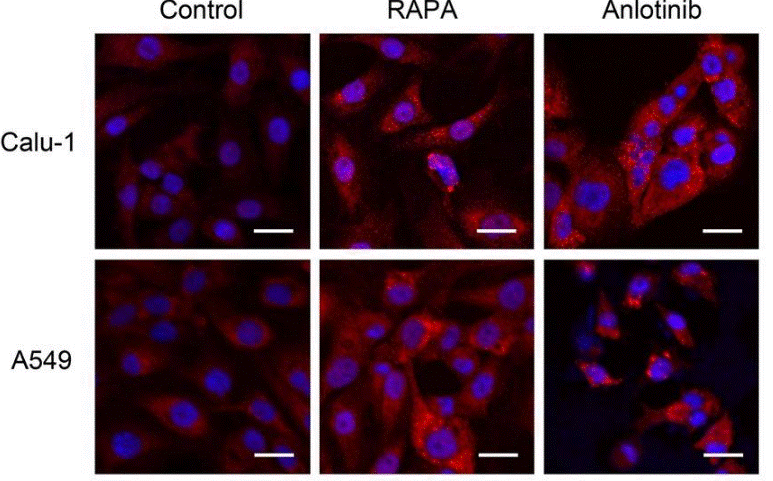
|
30755242 |
| Growth inhibition assay | Cell viability |
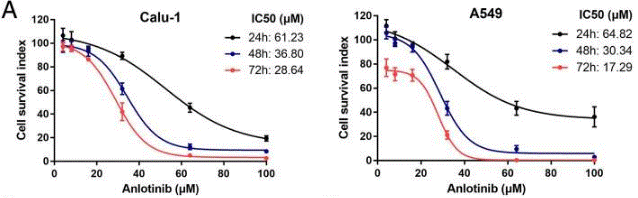
|
30755242 |
Informations sur lessai clinique
(données de https://clinicaltrials.gov, mis à jour le 2024-05-22)
| Numéro NCT | Recrutement | Conditions | Sponsor/Collaborateurs | Date de début | Phases |
|---|---|---|---|---|---|
| NCT06331169 | Not yet recruiting | Breast Cancer |
Fudan University |
April 1 2024 | Phase 1 |
| NCT06015061 | Recruiting | Pheochromocytoma Metastatic|Ultrasonography|Paraganglioma Malignant|Pheochromocytoma Malignant |
Peking Union Medical College Hospital |
March 1 2023 | -- |
| NCT05816499 | Recruiting | NSCLC Stage IV|NSCLC Stage IIIB|NSCLC Stage IIIC |
Shanghai Chest Hospital|The Affiliated Hospital of Qingdao University|Anhui Provincial Hospital|Zhejiang University |
February 16 2023 | Phase 1|Phase 2 |
| NCT05883085 | Recruiting | Pheochromocytoma|Paraganglioma |
Peking Union Medical College Hospital |
May 1 2022 | Phase 2 |
| NCT05301764 | Recruiting | Soft Tissue Sarcoma |
Lyvgen Biopharma Holdings Limited |
May 25 2022 | Phase 1|Phase 2 |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Non destinés à lusage humain. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.