réservé à la recherche
Tariquidar (XR9576) P-gp inhibiteur
N° Cat.S8028
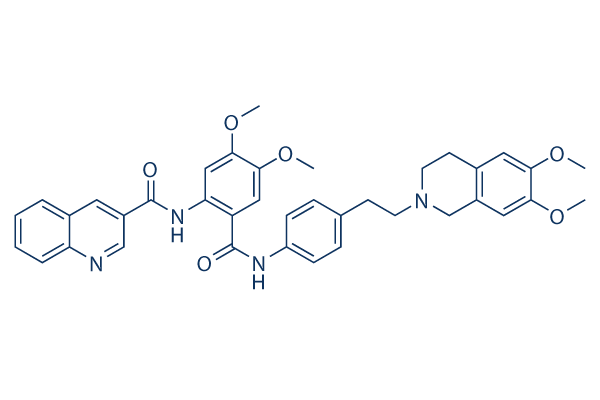
Structure chimique
Poids moléculaire: 646.73
Contrôle qualité
| Cibles apparentées | CFTR CRM1 CD markers AChR Calcium Channel Sodium Channel Potassium Channel GABA Receptor TRP Channel ATPase |
|---|---|
| Autre P-gp Inhibiteurs | Zosuquidar 3HCl Elacridar (GF120918) (20S)-Protopanaxadiol Solamargine Sinapine Levistilide A Evodine Valspodar Wilforine Encequidar (HM30181) |
Culture cellulaire, traitement et concentration de travail
| Lignées cellulaires | Type dessai | Concentration | Temps dincubation | Formulation | Description de lactivité | PMID |
|---|---|---|---|---|---|---|
| K562/DOX | Function assay | 1 uM | 10 mins | Inhibition of P-gp in human K562/DOX cells assessed as increase in rhodamine-123 efflux in human K562 cells at 1 uM incubated for 10 mins hrs by flow cytometry relative to untreated control | 28113128 | |
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| KB-V1 | Function assay | 200 nM | Inhibition of P-gp in human KB-V1 cells assessed as increase in rhodamine 123 accumulation at 200 nM | 21657271 | ||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| HepG2 | Cytotoxicity assay | 48 hrs | Cytotoxicity against adriamycin-resistant human HepG2 cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=37.2μM | 27328029 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells after 48 hrs by MTT assay, IC50=31.56μM | 28645831 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=31.56μM | 29631786 | ||
| A2780adr | Function assay | 10 uM | 30 mins | Inhibition of ABCB1 in human A2780adr cells assessed as increase in accumulation of calcein AM at 10 uM preincubated for 30 mins followed by calcein AM addition measured every 60 secs for 60 mins by fluorescence assay relative to control | 29547272 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells overexpressing P-gp assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=27.19μM | 29631786 | ||
| CCD-18Co | Cytotoxicity assay | 48 hrs | Cytotoxicity against human CCD-18Co cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| SW620/AD300 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620/AD300 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| HLF | Cytotoxicity assay | 48 hrs | Cytotoxicity against HLF cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=16.69μM | 27328029 | ||
| CEM/VLB500 | Growth inhibition assay | 3 days | Growth inhibition of human CEM/VLB500 cells after 3 days by resazurin assay, GI50=13.5μM | 17399990 | ||
| MCF7/ADR | Cytotoxicity assay | 48 hrs | Intrinsic cytotoxicity against human MCF7/ADR cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=13.1μM | 27328029 | ||
| HCT116 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human HCT116 cells assessed as cell viability after 48 hrs by MTT assay, IC50=12.5μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 12 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=8.28μM | 28645831 | ||
| KBV | Function assay | 5 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 5 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=5.24μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 6 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=4.97μM | 28645831 | ||
| KBV | Function assay | 10 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 10 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=4.46μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured immediately by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=3.02μM | 28645831 | ||
| K562/A02 | Function assay | 5 uM | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 at 5 uM measured after 48 hrs by MTT assay (Rvb = 43.75 to 96.91 uM), IC50=1.97μM | 28645831 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 measured after 48 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=1.6μM | 28645831 | ||
| MCF-7 MX | Function assay | Inhibition of BCRP expressed in MCF-7 MX cells using Hoechst 33342 staining, IC50=1.5μM | 21354800 | |||
| MCF7 | Function assay | Inhibition of ABCG2 in human mitoxantrone-resistant MCF7 cells by Hoechst 33342 assay, IC50=1.44544μM | 18678495 | |||
| HFE | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HFE cells assessed as cell viability after 72 hrs by MTT assay, IC50=1.28μM | 26197160 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells using Hoechst 33342 staining, IC50=0.94μM | 21354800 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 overexpressed in human MCF7/Topo cells by flow cytometric-based mitoxantrone efflux assay, IC50=0.916μM | 19170519 | |||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 staining based fluorescence assay, IC50=0.526μM | 30128080 | ||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 microplate assay, IC50=0.526μM | 24900683 | ||
| MCF7/Topo | Function assay | Inhibition of ABCG2 expressed in human MCF7/Topo cells by Hoechst microplate assay, IC50=0.526μM | 21570282 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 in human MCF7/Topo cells by Hoechst 33342 assay, IC50=0.52μM | 26774038 | |||
| KBV1 | Function assay | 10 mins | Inhibition of ABCB1 in human KBV1 cells after 10 mins by Calcein-AM microplate assay, IC50=0.223μM | 24900683 | ||
| Kb-V1 | Function assay | 10 mins | Inhibition of ABCB1 expressed in Kb-V1 cells after 10 mins by calcein-AM assay, IC50=0.223μM | 21570282 | ||
| KBv1 | Function assay | Inhibition of ABCB1 overexpressed in human KBv1 cells by flow cytometric-based calcein-AM efflux assay, IC50=0.223μM | 19170519 | |||
| KBV1 | Function assay | Inhibition of ABCB1 in human KBV1 cells assessed as inhibition of calcein-AM efflux, IC50=0.22μM | 26774038 | |||
| A2780 | Function assay | 30 mins | Inhibition of human Pgp in A2780 cells after 30 mins by Hoechst 33342 assay, IC50=0.12589μM | 18083034 | ||
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human KB-3-1 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.15 +/- 0.04 uM), IC50=0.11μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human OVCAR8 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.12 +/- 0.03 uM), IC50=0.08μM | 27504669 | |
| A2780/ADR | Function assay | Inhibition of P-glycoprotein-mediated multidrug resistance in adriamycin-resistant human A2780/ADR cells by calcein AM assay, IC50=0.078μM | 19250834 | |||
| A2780adr | Function assay | Inhibition of P-gp expressed in A2780adr cells by calcein AM accumulation assay, IC50=0.08μM | 21354800 | |||
| A2780 | Function assay | Inhibition of P-gp in human adriamycin-resistant A2780 cells by Hoechst 33342 assay, IC50=0.07244μM | 18678495 | |||
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.07 +/- 0.19 uM), IC50=0.07μM | 27504669 | |
| CEM/VLB500 | Function assay | 3 days | Reversal of P-gp-mediated multidrug resistance to in human CEM/VLB500 cells after 3 days by resazurin assay, EC50=0.068μM | 17399990 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.06457μM | 18083034 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.064μM | 18083034 | ||
| MDCK | Function assay | 30 mins | Inhibition of P-glycoprotein (unknown origin) expressed in MDCK cells assessed as reduction of calcein-AM transport after 30 mins by fluorescence assay, EC50=0.044μM | 24607999 | ||
| MDCK | Function assay | 30 mins | Activity at MDR1 (unknown origin) expressed in MDCK cells using calcein AM as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.044μM | 23374872 | ||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 3714.80 +/- 383.58 nM), IC50=0.01851μM | 27504669 | |
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 277.68 +/- 56.61 nM), IC50=0.00066μM | 27504669 | |
| HEK293 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against HEK293 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 5.28 +/- 0.74 nM), IC50=0.00495μM | 27504669 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells after 48 hrs by MTT assay, IC50=27.19μM | 28645831 | ||
| SW620 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 24 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=14.39μM | 28645831 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells by pheophorbide A assay, IC50=0.85μM | 19932960 | |||
| MCF7 MX | Function assay | Inhibition of BCRP expressed in MCF7 MX cells by Hoechst 33342 staining, IC50=0.68μM | 19932960 | |||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.54 +/- 0.60 uM), IC50=0.24μM | 27504669 | |
| MDCK | Function assay | Inhibition of MDR1 expressed in MDCK cells using rhodamine 123 staining by flow cytometry, IC50=0.21μM | 21354800 | |||
| CCRF-CEM/VCR1000 | Function assay | 240 secs | Inhibition of P-glycoprotein-mediated daunorubicin efflux from human CCRF-CEM/VCR1000 cells after 240 secs by FACS flow cytometric analysis, IC50=0.03311μM | 22452412 | ||
| HEK293 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 transfected in HEK293 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 504.65 +/- 44.94 nM), IC50=0.02477μM | 27504669 | |
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human KB-3-1 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.78 +/- 0.27 nM), IC50=0.00041μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human OVCAR8 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 8.53 +/- 1.95 nM), IC50=0.00518μM | 27504669 | |
| MDCK | Function assay | 30 mins | Activity at BCRP (unknown origin) expressed in MDCK cells using rhodamine 123 as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.01μM | 23374872 | ||
| HepG2 | Function assay | 10 uM | 90 mins | Inhibition of P-gp mediated efflux in adriamycin-resistant human HepG2 cells assessed as intracellular rhodamine-123 accumulation at 10 uM incubated in dark condition for 90 mins by flow cytometry relative to control | 27328029 | |
| Cliquez pour voir plus de données expérimentales sur les lignées cellulaires | ||||||
Informations chimiques, stockage et stabilité
| Poids moléculaire | 646.73 | Formule | C38H38N4O6 |
Stockage (À partir de la date de réception) | |
|---|---|---|---|---|---|
| N° CAS | 206873-63-4 | Télécharger le SDF | Stockage des solutions mères |
|
|
| Synonymes | XR9576 | Smiles | COC1=C(C=C2CN(CCC2=C1)CCC3=CC=C(C=C3)NC(=O)C4=CC(=C(C=C4NC(=O)C5=CC6=CC=CC=C6N=C5)OC)OC)OC | ||
Solubilité
|
In vitro |
DMSO
: 8 mg/mL
(12.36 mM)
Water : Insoluble Ethanol : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Entrez les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Entrez la formulation in vivo (Ceci nest que le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuiteμL PEG300, mélanger et clarifier, ajouter ensuiteμL Tween 80, mélanger et clarifier, ajouter ensuite μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuite μL Huile de maïs, mélanger et clarifier.
Remarque : 1. Assurez-vous que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue lors de lajout précédent est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
P-gp
(CHrB30 cells) 5.1 nM(Kd)
|
|---|---|
| In vitro |
Tariquidar présente une liaison de haute affinité à la P-gp avec une Bmax de 275 pmol/mg. Ce composé montre une interaction non compétitive avec les substrats de la P-gp. Il augmente l'accumulation à l'état d'équilibre de ces cytotoxiques dans les cellules CHrB30 à des niveaux observés dans les cellules AuxB1 n'exprimant pas de P-gp avec une CE50 de 487 nM. Cette substance chimique est capable d'inhiber l'activité ATPasique de la P-gp sensible au vanadate de 60 à 70%, avec des valeurs de CI50 puissantes de 43 nM. Il peut inhiber d'autres mécanismes de résistance à des concentrations plus élevées. 1 μM de ce composé abroge la résistance médiée par ABCG2 (BCRP) aux camptothécines in vitro. Il potentialise la cytotoxicité de plusieurs médicaments, y compris la doxorubicine; une inversion complète de la résistance est obtenue en présence de 25 à 80 nM de ce produit chimique. Dans MC26, une lignée cellulaire de carcinome du côlon murin avec une chimiorésistance intrinsèque, la CI50 de la doxorubicine est cinq fois inférieure en présence de 0,1 μM de ce composé (36 vs 7 nM). Dans les lignées cellulaires de carcinome mammaire murin, de carcinome pulmonaire à petites cellules humain et de carcinome ovarien humain avec une résistance chimithérapeutique acquise (EMT6/AR1.0, H69/LX4 et 2780 AD), la CI50 in vitro de la doxorubicine est 22 à 150 fois inférieure en présence de 0,1 μM de ce produit chimique. L'inhibition de la P-gp persiste pendant 23 h après son retrait du système de culture. Il a restauré la cytotoxicité de la doxorubicine dans le modèle de sphéroïde tumoral multicellulaire National Cancer Institute (NCI)/ADRRES dérivé de la lignée cellulaire de cancer du sein MCF7WT. |
| Kinase Assay |
Test d'accumulation de médicaments à l'état d'équilibre
|
|
Les cellules sont incubées dans un volume de réaction de 1 mL pendant 60 min à 37 ℃ sous 5 % de CO2 afin d'atteindre l'état d'équilibre. L'effet des modulateurs XR9576 sur l'accumulation de [3H]-ligand est étudié dans la plage de concentration 10-9 - 10-6 M. Ce composé est ajouté à partir d'une solution mère de DMSO donnant une concentration finale de solvant de 0,2 % (v/v). Après la récolte des cellules, le médicament accumulé est mesuré par comptage par scintillation liquide et normalisé pour la teneur en protéines cellulaires. Les tracés de la quantité accumulée en fonction de la concentration du modulateur sont ajustés avec l'équation générale dose-réponse : Y={(a-b)/(1+(X/c)d)}+bOù : Y=réponse ; a=réponse initiale ; b=réponse finale ; c=concentration EC50 ; d=valeur de la pente ; X=concentration du médicament.
|
|
| In vivo |
Tariquidar (2-8 mg/kg p.o.) s'est avéré potentialiser significativement l'activité antitumorale de la doxorubicine (5 mg/kg, i.v.) contre le carcinome murin du côlon MC26 in vivo. Dans les xénogreffes de carcinome humain, la coadministration de ce composé (6-12 mg/kg p.o.) a entièrement restauré l'activité antitumorale contre deux xénogreffes de tumeurs humaines MDR hautement résistantes (2780AD, H69/LX4) chez des souris nues. |
Références |
|
Applications
| Méthodes | Biomarqueurs | Images | PMID |
|---|---|---|---|
| Immunofluorescence | MRP7 |
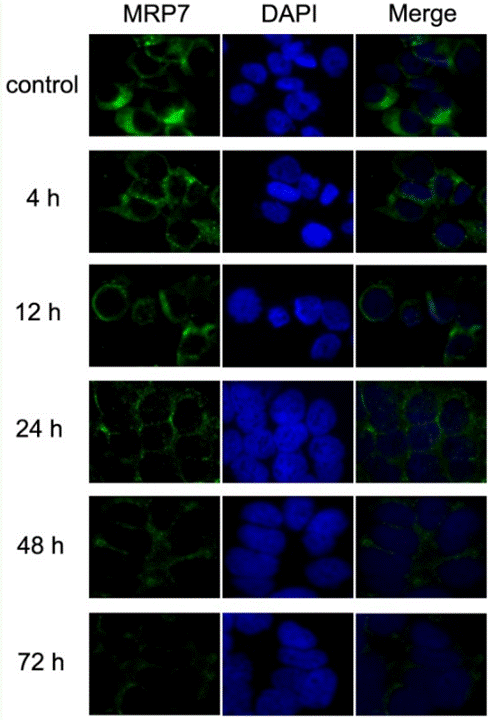
|
23393594 |
Informations sur lessai clinique
(données de https://clinicaltrials.gov, mis à jour le 2024-05-22)
| Numéro NCT | Recrutement | Conditions | Sponsor/Collaborateurs | Date de début | Phases |
|---|---|---|---|---|---|
| NCT01663545 | Completed | Epilepsies Partial |
National Institute of Neurological Disorders and Stroke (NINDS)|National Institutes of Health Clinical Center (CC) |
July 31 2012 | -- |
| NCT01547754 | Terminated | HIV-Associated Cognitive Motor Complex |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
January 9 2012 | -- |
| NCT01386476 | Completed | Drug Resistance |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
June 15 2011 | -- |
| NCT00082368 | Completed | Cancer |
National Cancer Institute (NCI)|National Institutes of Health Clinical Center (CC) |
May 16 2004 | Phase 2 |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Questions fréquemment posées
Question 1:
Can you please give me more specific and detailed information of how to dissolve and use it (S8028) for in vivo studies?
Réponse :
It in 30% Propylene glycol, 5% Tween 80, 65% D5W at 30mg/ml will be a suspension or emulsion. If you are going to administrate this compound by oral gavage, it is fine. We also have test some vehicles for it for i.p injection, and it is soluble in 5% DMSO+45% PEG 300+ddH2O at 2mg/ml clearly. When preparing the solution, please dissolve it in DMSO clearly first, then add PEG. After they mixed well, then dilute with water.
Les produits sont destinés à la recherche uniquement. Non destinés à lusage humain. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.