réservé à la recherche
EED226 DNA/RNA Synthesis inhibiteur
N° Cat.S8496
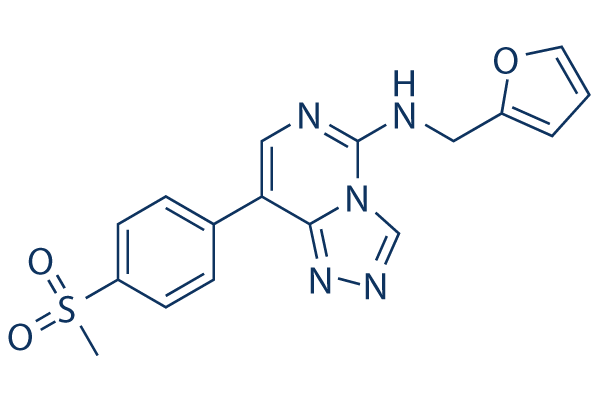
Structure chimique
Poids moléculaire: 369.40
Contrôle qualité
- Cité dans Nature Medicine pour sa qualité supérieure
- COA
- SDS
- Fiche technique
| Cibles apparentées | HDAC JAK BET Histone Methyltransferase PKC PARP HIF PRMT EZH2 AMPK |
|---|---|
| Autre DNA/RNA Synthesis Inhibiteurs | CX-5461 (Pidnarulex) B02 SCR7 Favipiravir (T-705) RK-33 BMH-21 Triapine (3-AP) Carmofur YK-4-279 Halofuginone |
Culture cellulaire, traitement et concentration de travail
| Lignées cellulaires | Type dessai | Concentration | Temps dincubation | Formulation | Description de lactivité | PMID |
|---|---|---|---|---|---|---|
| KARPAS422 | Antiproliferative assay | up to 14 days | Antiproliferative activity against human KARPAS422 cells harboring monoallelic Y641N EZH2 mutation assessed as reduction in cell viability measured every 3 to 4 days up to 14 days by Beckman Coulter-based method, IC50 = 0.08 μM. | 28092155 | ||
| G401 | Function assay | 48 hrs | Inhibition of EED in human G401 cells assessed as reduction in global H3K27me3 level after 48 hrs by ELISA, IC50 = 0.22 μM. | 28092155 | ||
| KARPAS422 | Antitumor assay | 300 mg/kg | 34 days | Antitumor activity against human KARPAS422 cells xenografted in Balb/C nude mouse assessed as tumor regression at 300 mg/kg, po BID for 34 days | 28092155 | |
| KARPAS422 | Antitumor assay | 1.5 to 40 mg/kg | 2 weeks | Antitumor activity against human KARPAS422 cells xenografted in Balb/C nude mouse assessed as reduction in tumor volume at 1.5 to 40 mg/kg, po BID for 2 weeks | 28092155 | |
| Cliquez pour voir plus de données expérimentales sur les lignées cellulaires | ||||||
Informations chimiques, stockage et stabilité
| Poids moléculaire | 369.40 | Formule | C17H15N5O3S |
Stockage (À partir de la date de réception) | |
|---|---|---|---|---|---|
| N° CAS | 2083627-02-3 | Télécharger le SDF | Stockage des solutions mères |
|
|
| Synonymes | N/A | Smiles | CS(=O)(=O)C1=CC=C(C=C1)C2=CN=C(N3C2=NN=C3)NCC4=CC=CO4 | ||
Solubilité
|
In vitro |
DMSO
: 73 mg/mL
(197.61 mM)
Water : Insoluble Ethanol : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Entrez les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Entrez la formulation in vivo (Ceci nest que le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuiteμL PEG300, mélanger et clarifier, ajouter ensuiteμL Tween 80, mélanger et clarifier, ajouter ensuite μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuite μL Huile de maïs, mélanger et clarifier.
Remarque : 1. Assurez-vous que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue lors de lajout précédent est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
EED
82 nM(Kd)
PRC2
114 nM(Kd)
|
|---|---|
| In vitro |
EED226 induit un changement conformationnel lors de la liaison à EED, entraînant une perte d'activité de PRC2. Ce composé inhibe également efficacement le PRC2 contenant une protéine EZH2 mutante résistante aux inhibiteurs compétitifs de SAM. Il régule la méthylation de l'histone H3K27 et l'expression des gènes cibles de PRC2 dans les cellules. Lors des essais enzymatiques in vitro, ce produit chimique inhibe le PRC2 avec une IC50 (concentration inhibitrice semi-maximale) de 23,4 nM lorsque le peptide H3K27me0 est utilisé comme substrat et une IC50 de 53,5 nM lorsque le mononucléosome est utilisé comme substrat, avec le H3K27me3 stimulant ajouté à 1 × Kact (1,0 μM). Il est non compétitif avec le SAM et le substrat peptidique. Ce composé se lie à EED et au complexe PRC2 avec une stœchiométrie de 1:1 et des Kd de 82 nM et 114 nM, respectivement. Il ne perturbe pas le complexe PRC2 et peut toujours occuper sa poche de liaison avec un inhibiteur EZH2 compétitif du SAM lié au PRC2. Ce produit chimique montre une sélectivité remarquable pour le complexe PRC2 par rapport à 21 autres protéines méthyltransférases, kinases et autres classes de protéines. La seule autre Histone Methyltransferase qui peut être inhibée par lui est le complexe EZH1-PRC2. Il, avec une perméabilité modérée, entraîne une diminution dose-dépendante des marqueurs H3K27me3 et H3K27me2 globaux dans les cellules G401.
|
| In vivo |
Ce composé induit efficacement la régression tumorale dans un modèle de xénogreffe de souris. Sa formulation en dispersion solide est bien tolérée chez les animaux. Ce produit chimique démontre clairement une efficacité dose-dépendante dans l'étude de xénogreffe de souris. Il inhibe la croissance des xénogreffes de lymphome diffus à grandes cellules B (DLBCL) et réduit les niveaux de H3K27me3 dans une mesure similaire à celle d'un inhibiteur d'EZH2. Il présente une très faible clairance in vivo et in vitro et une biodisponibilité orale d'environ 100 %, un faible volume de distribution (0,8 L/kg), une t1/2 terminale raisonnable (2,2 h) et une liaison modérée aux protéines plasmatiques (PPB) (14,4 %). Sa solubilité est relativement faible et peu dépendante du pH du milieu.
|
Références |
|
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Non destinés à lusage humain. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.